Also note that tweets during his talk have been collected by ONT into a Twitter Moment
Accuracy: Training Sets
After the obligatory introductory material (and remarking "Keith, we'll get the version number bit sorted out"), Clive spent a big chunk at the beginning talking about different approaches to accuracy. He noted that there are four major areas which ONT can work on to increase accuracy: Training Sets, Base Calling Algorithms, Assembly and Polishing and Chemistry.
Clive mentioned that they've realized their training sets weren't sufficient. I've worried for years that ONT was relying on lambda phage for training. Lambda has a very middle-of-the-road GC content and is completely missing some short GC-rich kmers (I looked a long time ago; I think it doesn't have every 6-mer). Given that I work (for at least a bit longer) almost entirely with organisms that are around 70% GC, this has always concerned me. Something small like lambda might have almost made sense when nanopore had low throughput, but with MinION getting 10s of gigabases and PromethION 100s of gigabases, that makes no sense any more. Actually better alternatives have existed even in those days: three years ago I made the suggestion of synthesizing specific test articles covering greater sequence space. Realistically, once MinIONs started generating multiple gigabases per run a cocktail of bacteria -- say Bacillus subtilus to cover low-GC, E.coli for mid-GC and Streptomyces coelicolor for high-GC -- would work nicely. Well, it may be awfully late, but at least they recognize the potential problem.
Accuracy: R10
Clive then moved on to R10. R10 is a new pore type and will be launched as a new flowcell in all three sizes (Flongle, MinION and PromethION) and seemed to suggest that these would be available to order in about two weeks with delivery in Q1 2019. My guessing of a mixed flowcell was completely off-base; apparently R9.x and R10 pores are incompatible (Clive said something about software, but didn't go into detail). Clive mentioned that a companion pore is being engineered to enable such mixes, but that is a ways off.
R10 has a longer base-contacting constriction in the pore, leading to far better homopolymer resolution than R9.4.1. One can get a sense of this from the plot below -- though the X-axis is clearly not aligned correctly. Also it is unfortunate it doesn't cover longer lengths; which (as Clive noted) most genomes have a lot of short homopolymers and few long ones, those few can be very important to some people (not me: long runs of G or C just aren't common, and forget about runs of A or T in Actinomycetes).

R10 has higher consensus accuracy than R9.4, and better yet the scores shown by Clive compare R10 polished with a fast polisher that works on base calls versus R9.4 with the slower (but excellent) nanopolish that runs on signals. For a depth of 25 this worked out to about +3 on the quality score, but for depth 150 it's about 8 phred points, hitting a Q-score of 42. Consensus accuracy for base identity approaches Q50.
Accuracy: Basecalling Algorithms
Clive switched into discussing basecallers next. Luckily he saved the picture of a dead tuna for the end -- the Albacore basecaller is being phased out immediately. In it's place are Flappy and Guppy; the former works on conventional processors whereas the latter requires GPUs.
Flappy generates results Clive compared to old ABI trace files: at each called position there is a probability for each base. It also uses what ONT calls a "flip-flop" algorithm to help with homopolymers -- in their model each base is represented by two different states (flip and flop aka + and -). Everything is flip (+), except in modeling homopolymers -- for the same base only flip-to-flop and flop-to-flip transitions (did ONT bioinformatics hire someone named Geisel?). Having these extra states somehow helps with the model and leads to better homopolymer performance.

An interesting side-effect of the flip-flop model is it easily accommodates additional states -- such as a state for methyl-C. These calls were shown to have high concordance with bisulphite data run on Illumina.
Flappy is available now from ONT's GitHub site; Guppy should be released by mid-December.
Clive also spoke of another approach to improved accuracy which they are calling 8B4. This adds a step to library production in which the native DNA goes through one round of synthesis with all four natural nucleotides but also four base analogs. The differences in signals in the unnatural analogs helps better elucidate homopolymers. I proposed this two years ago, getting some derision in the comments that it was turning the hard problem of calling four bases to the far worse problem of calling eight bases. In any case, ONT is refining this technique.
Overall, Clive believes the improvements shown could lead to consensus accuracy of Q60, or one error per megabase.
Accuracy: 1D^2
But that's not all. ONT is still plugging away on the 1D^2 chemistry, now claiming that 70% of first strands are followed by the complementary strand going through the pore. They've also built a better algorithm to combine the information from the two strands. Coming soon (supposedly 2018) are unique molecular identifiers to unambiguously pair the two strands, enabling use of 1D^2 with low complexity samples such as amplicons -- both the forward and reverse strand from the same molecule should have the same identifier. There will also be a 1D^2 kit for any PCR library approach and this apparently drives the 1D^2 efficiency up over 75%. Oxford is also claiming that for amplicons using their own unique molecular identifiers (UMIs) to identify the original templates, rare variants of less than 1% frequency can be reliably called by clustering reads on the UMIs prior to variant calling.
Accuracy: LCS
Oxford has yet another chemistry scheme for increasing accuracy which they are calling Linear Consensus Sequencing or LCS (the term was described as "cheeky" by Clive, with reference to some other platform's Circular Consensus Sequencing).
LCS uses some proprietary chemistry -- a sketch of which was covered by an incompletely opaque rectangle -- to convert inputs in the 5-15 kilobase range in to a set of linked copies of that that template. This is much like the output of rolling circle amplification (RCA), but Oxford's chemistry apparently never creates a circle. This is claimed to enable Q30 for the consensus of a single set of such duplicates (i.e. all from the same input molecule) and slightly lower for R9.4.

Accuracy: Fixed Issue with Low Complexity Regions
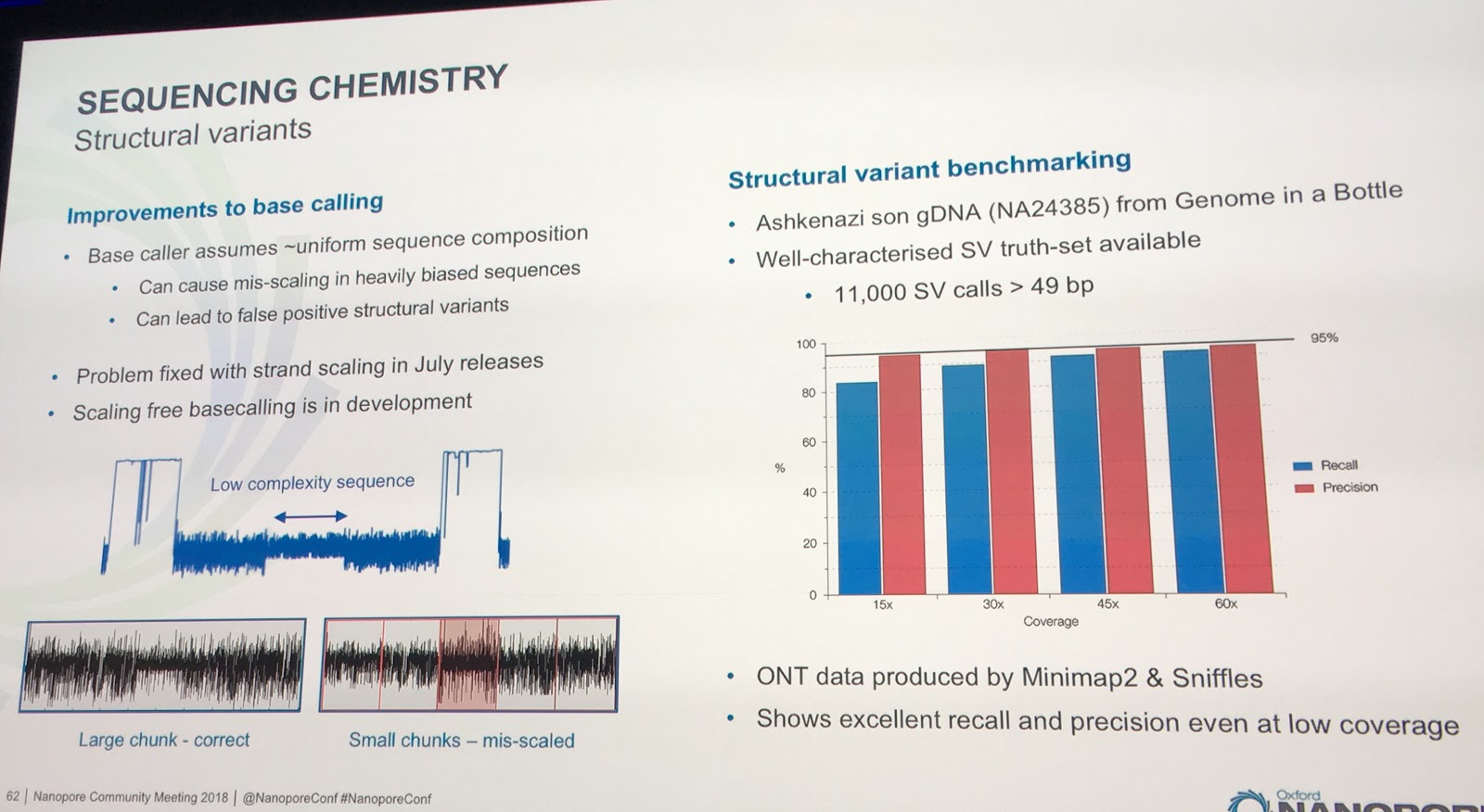
Clive also mentioned that a problem had been fixed which had resulted in false positive structural variant calls in low complexity regions. I wasn't familiar with the scaling terminology, but I think this ends up being an issue of needing to break the signal up into chunks for practical calling and then being unsure how to stitch the chunks together again. Anyway, now fixed
Enrichment
Okay, done with accuracy. Clive mentioned the very clever Cas9 enrichment scheme, which is detailed in an ONT poster. Having worked on a similar problem, I had to slap my head in amazement. The protocol takes only 90 minutes, but yields such high enrichment that one can get high-depth coverage on multiple human targets using only a Flongle! The key trick is to dephosphorylate the ends after initial DNA purification; this inactivates them for adapter ligation. Cas9 is used to target the regions of interest, cutting adjacent to the target using user-specified guide RNAs. Cutting -- and also activating for ligation! I must drop into curmudgeon mode and say never plot this way -- each figure in a trellis with the same Y-axis units should have the same bounds unless you have a damn good reason to do otherwise -- and none such exists here. Clive did not talk at all about the curious shoulders on the ROI regions of these plots, nor were off-target rates discussed -- the focus was on the fact that high coverage could be obtained for targeted regions.

I haven't had a chance to try these -- the fact that bacteria don't polyadenylate their mRNA makes the direct RNA tricky (and I heard at dinner from someone trying to adapt to this by artificially polyadenylating the bacterial RNA -- apparently it's harder to get working than it sounds). Direct RNA and Direct cDNA (PCR-free) are now enabled on PromethION and both have higher throughput (not described in detail for RNA; over 2X greater output for Direct cDNA). PCR cDNA has also been enabled on PromethION with about 20% greater throughput and an input amount dropped from 50 to 1 nanogram.
Throughput
Clive is always promising greater throughput, and tonight was no exception. In ONT's laboratories, the old "RevC" flowcells give up to 20 gigabases per run and "RevD" up to 30 gigabases; in ONT's R&D they've hit almost 50 gigabases. Now, in the field the best RevD runs seem to be around 20 gigabases and most people get 10 gigabases, so discount accordingly. A new electrochemical mediator ("3:1" -- unclear what those numbers mean), a new membrane and an R9 mutant are the drivers of improvement. Somewhere along the way Clive mentioned that in the current chemistries adapted DNAs that haven't found a pore yet still burn ATP "fuel" and this fuel consumption hurts performance late in runs; Oxford is working to reduce this useless idling.
For PromethION, median field output is now 60 gigabases and some users are semi-regularly getting 100 gigabases and a field site has hit 148 gigabases in one run. In contrast ONT is regularly getting 200 gigabases and has hit 220 -- and Clive thinks 300 will be topped soon. He showed data suggesting that field users tend to catch up to ONT in about six weeks.
I talked to two users and both said that PromethION flowcell supply is now reliable, so apparently I waited to long to report what I had heard that they weren't!
The display area has the new look for PromethION, a slickly designed box. The old single door to the flowcells has been replaced with six individual doors accessing one row of eight flowcells. Well, that's in the PromethION 48 -- Clive announced that they will offer 24-cell and 48-cell versions of the system. Pricing for these and for a new scheme for flowcells -- rather than making a long term commitment with scheduled shipments one gets a single drop of flowcells warrantied for 3 months -- were presented. I've routinely misinterpreted these, so I won't even attempt it here. The production PromethIONs are slated to go out "Q2 2019", with ordering starting in January.

Further Throughput Improvements
Clive dangled several further improvements for 2019 and beyond. ONT is working on improving the duty cycle -- i.e. reducing the amount of time a pore is idle between templates. Increased sensitivity would mean lower amounts of input DNA. Reducing the idle ATP burn I mentioned above. The possibility of 1000 bases per second -- rather than current 450 - -is apparently again in the air. And further improvements to basecallers.
Clive also brought up switching to Field Effect Transistors (FETs) in place of the current ASICS. FETs would potentially enable packing three times as many pores per device, with a similar increase in throughput.
Clive exuded confidence at this point -- "they will never catch up! - and you know who they are!"
MinION Mk1C & MinIT
In the just-before-conference-start (actually composed at breakfast) piece I guessed wrong on Clive's teaser tweet: what he had shown was the new look for the MinION Mk1C. This device has all of the necessary hardware -- a MinION, the MinIT compute system and a touchscreen to control it all. Actually, it has a bit of everything -- WiFi, GSM/UMTS/LTE cellular support, 500 gigabases of onboard storage, an SD slot and an NVIDIA GPU.

ONT plans to ship Mk1C devices mid next year. $5K will get you a device, 6 flow cells and a library kit -- but with the warranty lasting only as long as those flow cells. $10K gets a device, 8 flow cells and 2 kits -- plus a three year warranty and software license.
I spoke with some ONT folks about the MinIT, and it's basically a little Linux box with GPUs in it. The main CPU is based on ARM, but many bioinformatics packages run just fine on this. MiniMap2 and NGMLR for example.
One concern: with the crazy performance improvements Clive is promising, that 450Gb SSD on these devices might start looking very small. Indeed, some PromethION users are already reporting that 12 flowcells running can tax network capacity (if only one of the two possible 10Gbit connections is available) and even petabytes of storage could disappear under a torrent of FAST5 files -- which ONT still has a raging debate on whether to replace or not.
Flongle
Clive was quite pleased with early Flongle reports. He mentioned his dentist would love to have one to type the bacteria causing tooth problems in patients. An unverified number puts 20 Flongles in the field. Clive's release summary slide says the program will expand in January, but no details were given.
VolTRAX v2
Clive admitted that VolTRAX has been a very delayed project (given he was promising full release in January 2016 and now release is slated for early next year, that's an understatement).
VolTRAX now has the capability to perform PCR, optical detection. Library multiplexing of up to 10 samples is now possible. But the first release will be only a 1D kit with multiplexing as an option. By multiplexing, the per-sample library cost should be $30.

Clive also talked about the sequencing-on-VolTRAX concept introduced at London Calling. In this scheme, pairs of droplets are used to form the two compartments with an intervening membrane and a pore is popped in. Apparently this is akin to an idea Hagen Bayley had back in 2005.
An interesting detail I haven't heard Clive drop before is that pores don't like to go into the membranes and must be driven in electrically. Since the addition of a pore changes the electrical properties, the current can be turned off -- which is how Oxford achieves super-Poisson (one and only one pore per membrane) loading of pores into their membranes.
Anyway, they've actually generated raw data and have a prototype (which Clive thinks is too big) and a name (VolTRAXion, which Clive doesn't like). Using this for single cell sequencing is the application that Clive likes best -- with cell lysis happening in droplets on the instrument.
Miscellaneous
Oxford is expanding the native barcode set from 12 to 24 -- long overdue, but still far too few barcodes. Ambient rapid kits are scheduled for January -- these have all reagents lyophilized.
As always -- see errors or want to challenge my interpretations? Let the comments fly!
3 comments:
Hi Keith,
Thanks for the speedy wrap up! Saved me wading into the tweets.
I wanted to propose an explanation for the shoulders you mentioned for the Cas9 enrichment technique. The approach uses two separate Cas9 RNPs containing (presumably) different guide RNAs, which will likely each cut with different efficiencies. If one RNP cuts better than the other, this would lead to some products with one digested end only. This would presumably still be a substrate for adapter ligation, but the read would not stop at the predicted position as the other RNP did not cut. This would explain the lop-sided shoulders for several of the sites tested (INS, SCA3, FMR1) - one RNP is much more efficient than the other in each case.
Shoulders on both sides could be explained by both guides having similar efficiencies, but both not cutting to completion, leaving some single-cut products to be ligated and sequenced from each side. This looks to be the case for the SCA2, SCA6 and HTT sites in the figure.
There are a few strange anomalies in the data still: at the MUC1 and INS sites the shoulders themselves seem to stop abruptly. If my explanation is correct, there should be a long, tapering tail that will depend on the natural, random fragmentation of the DNA used to prepare the input sample (as it looks to be at the FMR1 and SCA3 sites). So why the abrupt stops in the MUC1 and INS examples? Could these be due to off-target cut sites by the RNPs? Seems unlikely that they would occur so close to the intended cut sites. Could these be regions that break more often due to shearing? Is that a known phenomenon - non-random shearing? That's all I can come up with right now, but let me know if you can think of anything else!
Hope this makes some sense!
Cheers,
Mike D'Angelo
Hi Keith,
Thanks for this very useful summary, full of information. I have one query: the throghput you indicate for PromethIon (200 G at Oxford Nanopore) is for ONE PromethIon flowcell, not 24 or 48? Or am I hopelessly confused?
Thanks for clarifying,
Bertrand
Bertrand: a very unfortunate detail omitted by me! That’s 200Gbases per flowcell - so many terabases per instrument
Post a Comment