Counterr takes an indexed BAM file (I'm using minimap2 to generate mine) and the genome for that BAM and generates a large number of plots in a PDF report as well as some tab-delimited files for further analysis. In particular, there is a breakdown of context-specific error. I'm not going to look at that now but instead just focus on some high level statistics.
I'm going to show data based on a proprietary actinomycete genome with a characteristically high G+C content. We'll assume I've assembled and polished it correctly but I won't detail that process here. This data was generated with the Oxford platform in late autumn using the current version of R9.4.1 and MinKNOW (or it was current then).
At NCM Clive Brown declared the old Albacore basecaller to be dead, with a gruesome image of a dead tuna to emphasize the point. The new "flip-flop" basecalling regime is available as a research grade tool called Flappie right now, with a production grade tool called Guppie to be released very soon (perhaps next week?).
Installing Flappie seemed to find every little quirk in our Linux installation. First you must install Git LFS to deal with some of the large model files. Then I had a round of issues with finding shared object libraries to get it to compile. Then another round of shared object library issues with nodes on our cluster. But finally, it is working. Or should I say w...o...r...k...i...n...g -- I can confirm all the reports that Flappie is painfully slow. Guppy is supposed to be faster and will be GPU-enabled, so if you have GPUs or MinIT or can get GPUs in the cloud, I would definitely recommend exploring that.
Flappie doesn't lack for confidence in itself -- it sometimes gives bases phred quality scores of 50 -- which I don't at all believe. But let's look at some real data.
Homopolymers
Sequencing errors in long read technologies are heavily dominated by homopolymers. Given that this is a very G+C rich genome, we should not be surprised to find the genome rich in polyG/polyC and poor in polyA/polyT. As we'll see, there are a few length 6, 7 and 8 homopolymers, but they are rare -- and none are longer. Presumably this reflects pressure against long runs of G which might form quartet structures.


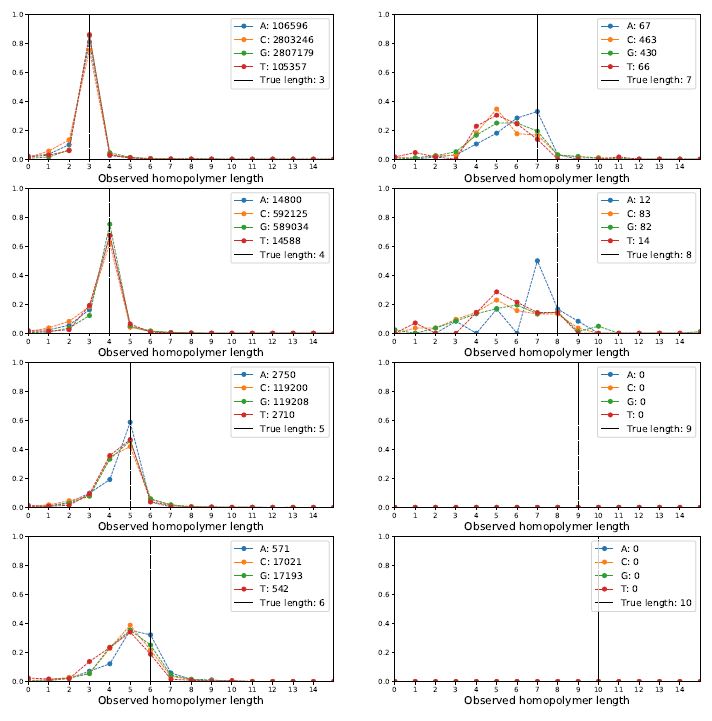
Okay, how does the Albacore-called data look?

We can see that for length=3 or 4 things look quite good -- the distribution has a sharp peak centered on the correct length. The shoulder for calling the array one too short is a bit bigger than one too long. But, as homopolymers grow to 5, 6, 7 and 8 the distributions get much uglier and not centered correctly.
I have a much smaller Flappie dataset due to the slow calling and working Flappie tests into a queue stuffed with production work, but we can get a peek.
The best improvement is in the length 5 homopolymers; now instead of being consistently low on these there is a sharpish peak centered on the correct value. But length 6 doesn't seem to have gotten much better and the length 7 is a cleaner distribution but centered badly. Length 8 is a tiny sample but has a horrid distribution.
Substitutions
Of course we are also interested in getting correctly called bases both inside and outside of homopolymers. If we just look at non-homopolymeric regions,here are the results of Albacore and Flappie side-by-side.

For Albacore and pure substitution errors, A miscalled as G dominates with G miscalled as A not far behind. As are also dropped out more frequently -- twice the rate of Ts and there is a higher probability of calling a phantom A (i.e. where no base should be) than anything else as well.
If we look at the Flappie results, there are a bunch of patterns going on. For bases that have been called, the miscalls are actually going up in every case -- sometimes spectacularly. C being miscalled as G has quadrupled! G as C as well as G has doubled and so has A as C! And many others. But missing bases all together has gone down significantly -- missed As by about a third for example. Excessive calls -- calling a base when none is present, hasn't changed radically but is in the correct direction.
Conclusions
I'm always suspicious of Oxford's training sets for their basecallers. I've long worried that they were mostly using lambda phage, which is very GC-neutral and missing a lot of important kmers in the length 5 and 6 range that is of great interest. It certainly lacks many of the long homopolymers in a variety of contexts. I've long advocated using synthetic standards designed to calibrate known issues, but even just using some large genomes with varied G+C could make a difference. I started to look at some E.coli data we have, but that was generated over a year ago using older chemistry and older MinKNOW, so not directly comparable.
I'm now very unsold on the advantages of the new basecaller scheme -- it makes some things better but some worse in my hands and its not clear there's an advantage overall -- and it certainly takes much longer. What I really need to do is feed all of this through my favorite error correction schemes or downstream applications to see what the overall advantage is (or isn't).
If you have data -- particularly data from genomes with biased nucleotide content -- I would encourage you to do similar analyses. And to dig through the mountain of other graphs and tables counterr produces that I didn't go through.
1 comment:
Hi Keith
Thx for the great blog post.
What GC did you organsim exactly have?
Best Michael
Post a Comment