Flowcell Priming & Loading
I've written of past travails with flowcell priming and loading. A session led by ONT's Cameron Knight in the middle covered this in a practical manne. There was a good live demonstration of appropriate technique, with nice magnified video feeds. Then we got to play with flowcells (we were both assured they were duds and asked to leave them behind at the end!).




Besides the fact I don't pipet for a living, I have a general challenge with fine motor skills. Sometimes this has involved creative solutions. When I was an undergrad taking quantitative chemistry, I was having miserable luck with titrations as all-too-frequently I would try to open the burette to drip in something and instead open it quite wide. Overshoot with the base followed by overshoot with the acid. Rising frustration no help there. What I finally learned to do was put one finger behind the toggle so that when I opened the valve I was jamming the toggle into my finger. This wasn't comfortable in the least, but that resistance made sure I didn't overshoot my mark.
Priming seems to mostly be a matter of not pushing past the first stop on the pipet Drawing out the bubble first is important, but a key lesson from the live demo is that there is a lot of leeway here in terms of how much buffer to draw. As long as the buffer front is drawn all the way to the sensor array, things are quite safe. Oh, and also correctly docking your tip with the port -- my first attempt ended with a lot of buffer on the surface of the device. So some warm-up with used flowcells would be in order until I really get this down!
Knight also mentioned a conservative technique for the bubble withdrawal -- use the pipet's volume adjustment wheel to slowly draw up buffer. Knight also mentioned the reason why the second 200ul flush with the SpotON port should be followed immediately with sample loading: otherwise a bubble may re-form at the SpotON port. And remember your eyes don't lie to you: the loading beads settle in no time at all, so be sure to gently re-mix them before and after adding running buffer to the loading mix.
The dripping in of the sample turns out to be easier than I thought, once I've seen it a few times. I did use a two-handed scheme of the pipet plunger so that each hand is delivering a little force; somehow that helps (at least psychologically) with my slow (but not too slow) delivery.
(image courtesy of Oxford Nanopore)
Library Quality
If priming and loading go well, then run quality is believed to be mostly driven by library quality. Library quality in turn appears to be largely driven by the amount and quality of sample DNA.
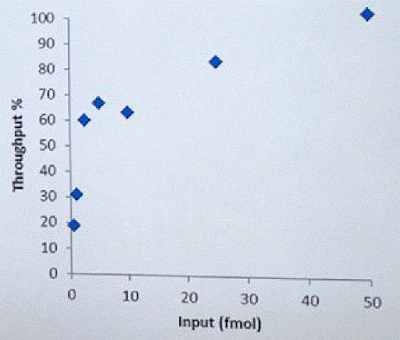
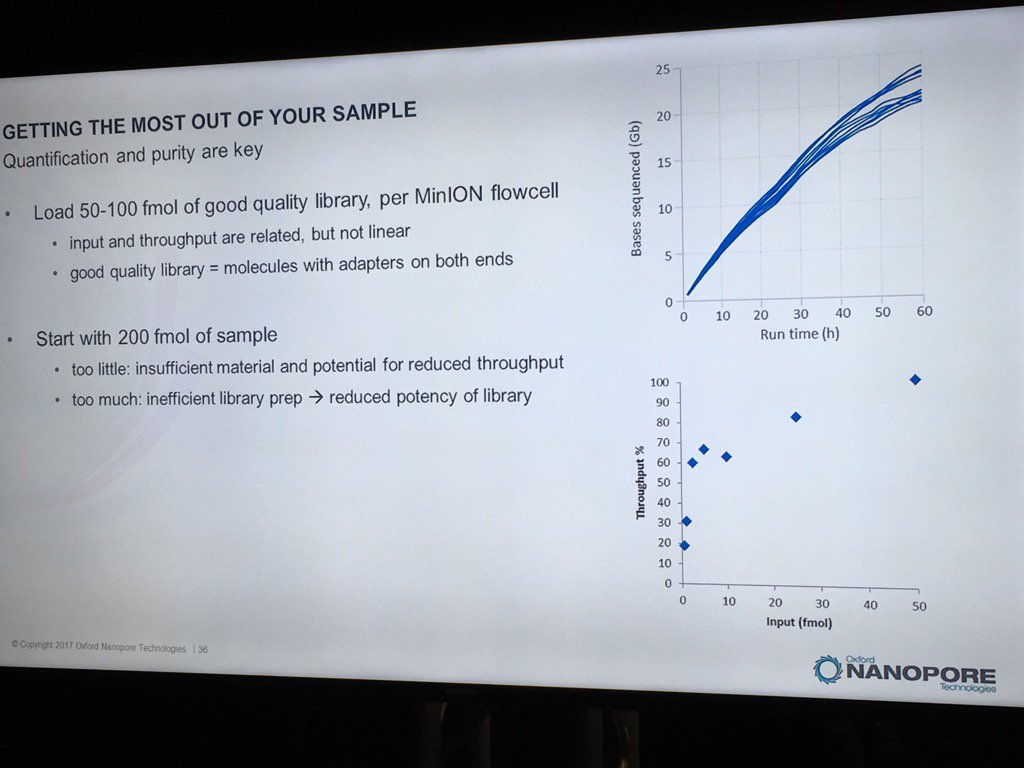
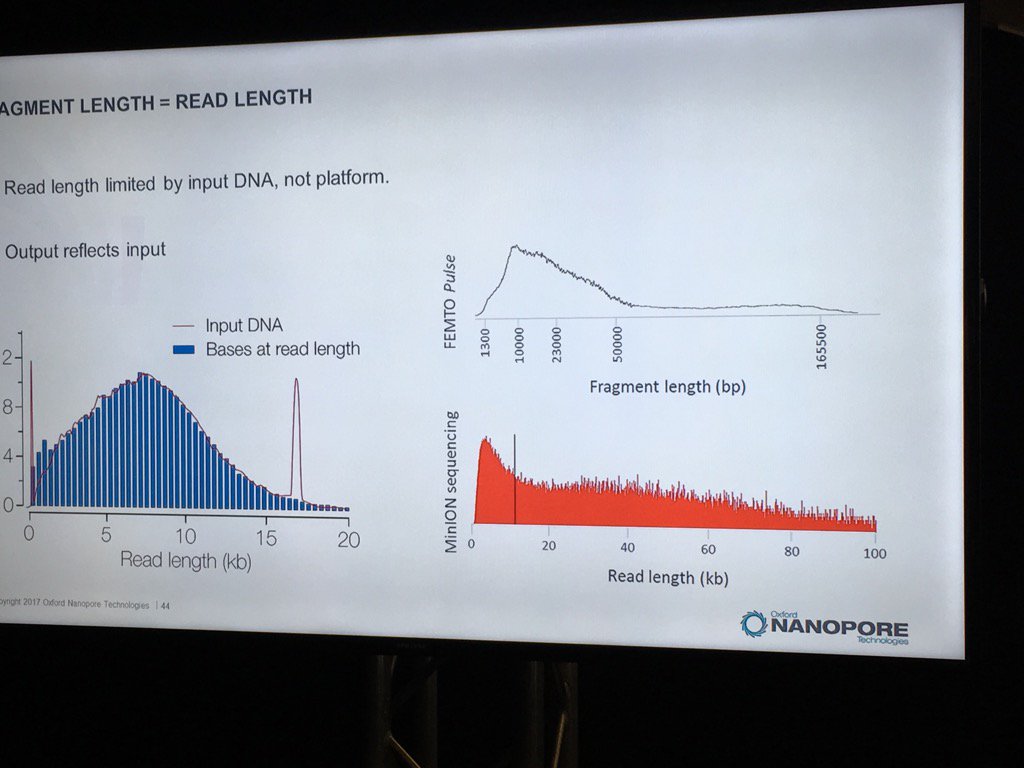
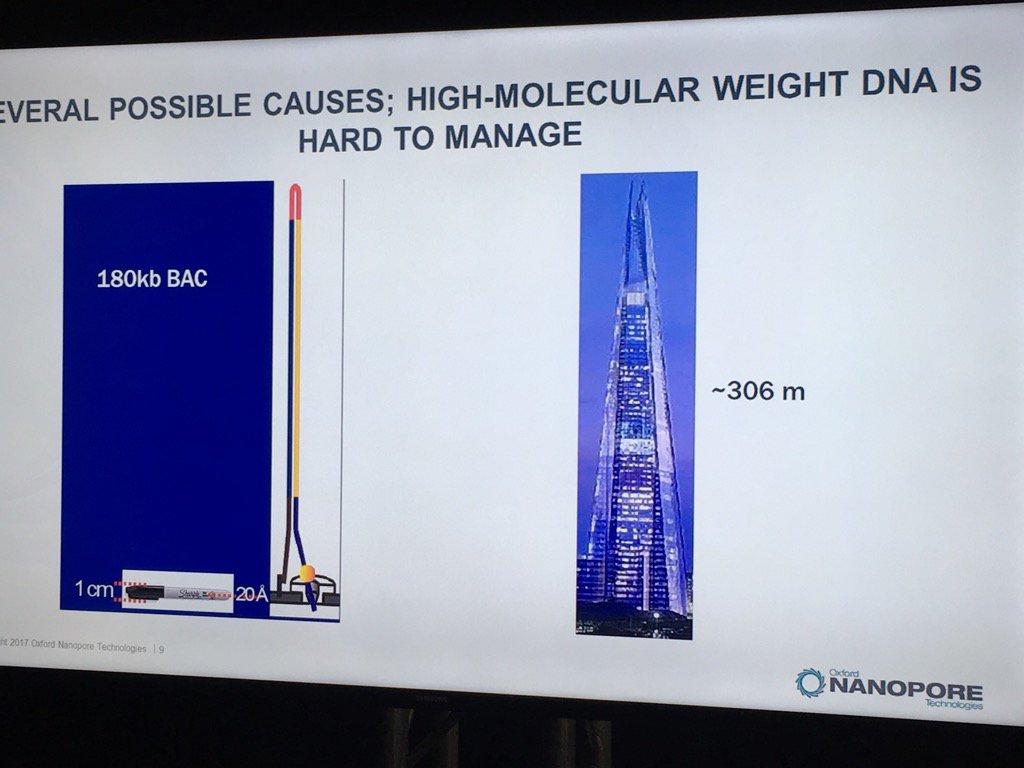
David Stoddard from ONT gave Oxford's current view of the situation. Looking back at this, their DNA input guidelines do suggest using a lot of DNA. -- 200fmol. That may not sound like much, but 200fmol of 10kb fragments = 1.32 ug of DNA (according to Promega's calculator and checked with another one). So for larger fragments, scale that amount accordingly. That can lead to some seemingly absurd input amounts -- 13ug for 100kb BACs. However, as the curve below shows, overall yield stays relatively high (~60-70%) with a tenth that amount of input DNA
An interesting tidbit that David mentioned is that Oxford has shown that molecules with adapters on both ends sequence much more readily than ones with adapters on only one end. They've proven this by constructing molecules with controlled addition of the loading adapter and/or tether at each end, and having two tethers appears to be very advantageous. I asked about how to measure the fraction of the library in the two adapter (and therefore two tether) state, and David said there isn't a good way. Ola Wallterman suggested via Twitter that this would be a useful stat for MinKNOW to report, which might even trigger a user to halt and try remaking the library. The two tethers does go someways to answering a question I've wondered about: what is the drawback to overloading the library reaction? For the ligation protocol, excess input DNA would probably result in a lower fraction of fragments adapted at both ends and therefore the fraction which carry tethers at both ends.
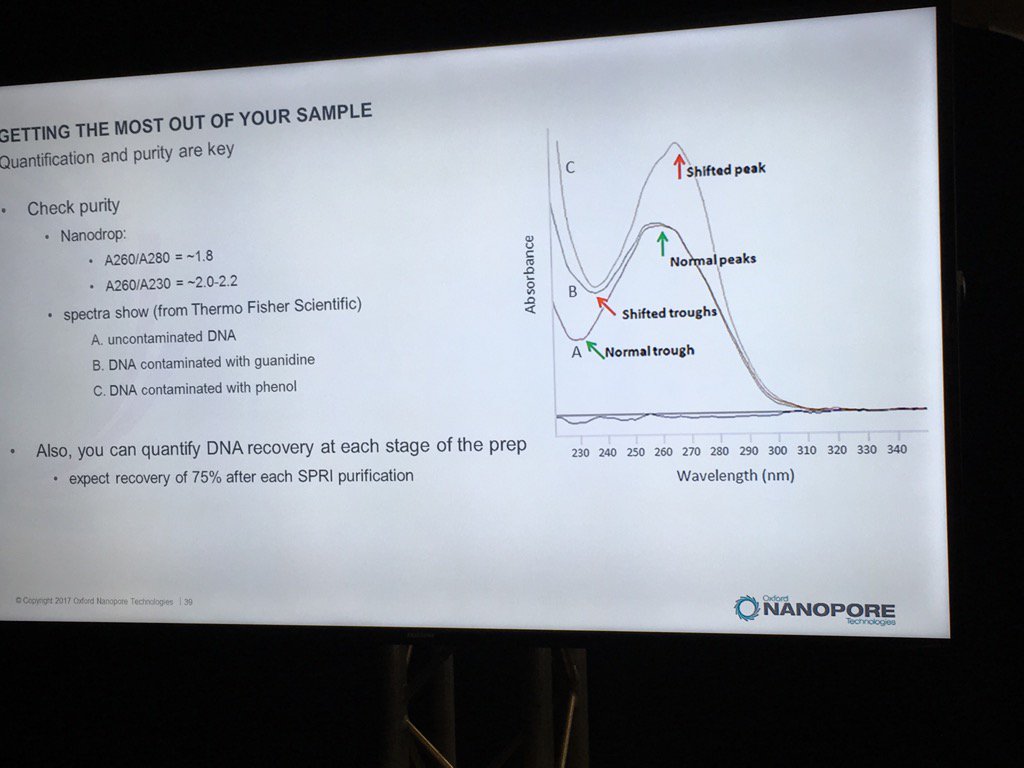
Contamination of DNA preps is always a concern, but what matters? We once dropped a short read project with one vendor after far too many back-and-forths about the RNA contamination in our sample. David thinks RNA is "benign", and they've done some limited spiking experiments to back that up. The catch is that RNA contamination may throw off DNA quantification, even with the more trusted Qubit (vs. Nanodrop) method. He believes the worst offenders are carryover phenol, guanidinium, detergents and perhaps carbohydrates. He also pointed out that with the Rapid 1D prep the original sample buffer is carried into the flowcell, whereas the ligation protocol (or short read libraries) involve bead cleanups and don't have much if any carryover. He recommends TE for the buffer.
For determining purity, David suggests going back to the old standbys of A260/A280 and A260/A230 ratios. A reminder, RNA contamination may throw DNA quantification off.
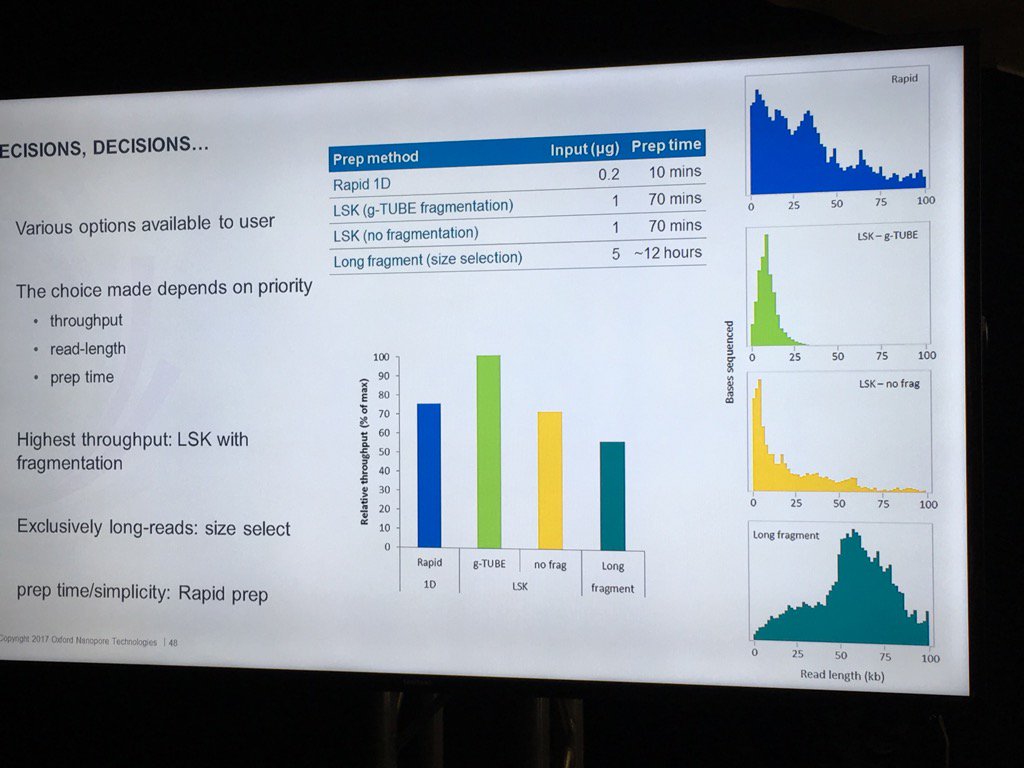
Stoddard also did a nice job of pointing out the trade-offs involved in choosing different library preps. For example, the Rapid 1D has a very short and easy protocol and gives long reads, but only about 70-80% as much data as the 1D Ligation kit. There are two low input kits that utilize PCR (see "PCR Solutions" screenshot below). Again, the rapid kit is transposase-based and is simpler, but exhibits shorter read lengths than the low input ligation-based kit. Another example: the direct RNA kit uses a 70bp/s motor vs. the 450bps motor for sequencing cDNA with the ligation kit. So if you want to avoid reverse transcription or see RNA modification, then direct RNA is clearly best, but the cDNA approach may make more sense if overall throughput is to be maximized. Armed with this information, users can choose a kit based on the most important properties for their specific experiment.
David also talked about input DNA size. ONT continues to be convinced that the pores will sequence the full length of DNA presented to them. Pulsed field is probably the best way to size HMW DNA, though two different pulsed field setups may give very different results. It was also commented that this is putting a complex, expensive technology upstream of a simple, inexpensive technology, so routine use of pulsed field for DNA characterization may not make sense.
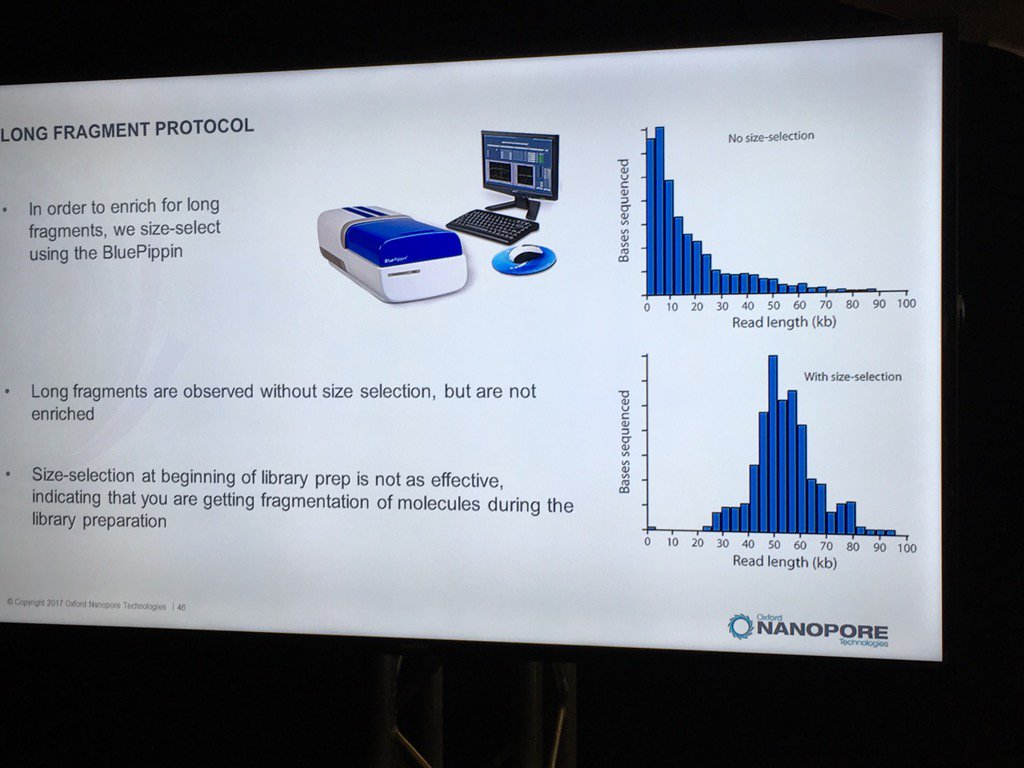
Blue Pippin sizing is a route to runs which are nearly only long reads, but this size selection is most effective applied to the completed library rather than immediately after fragmentation.
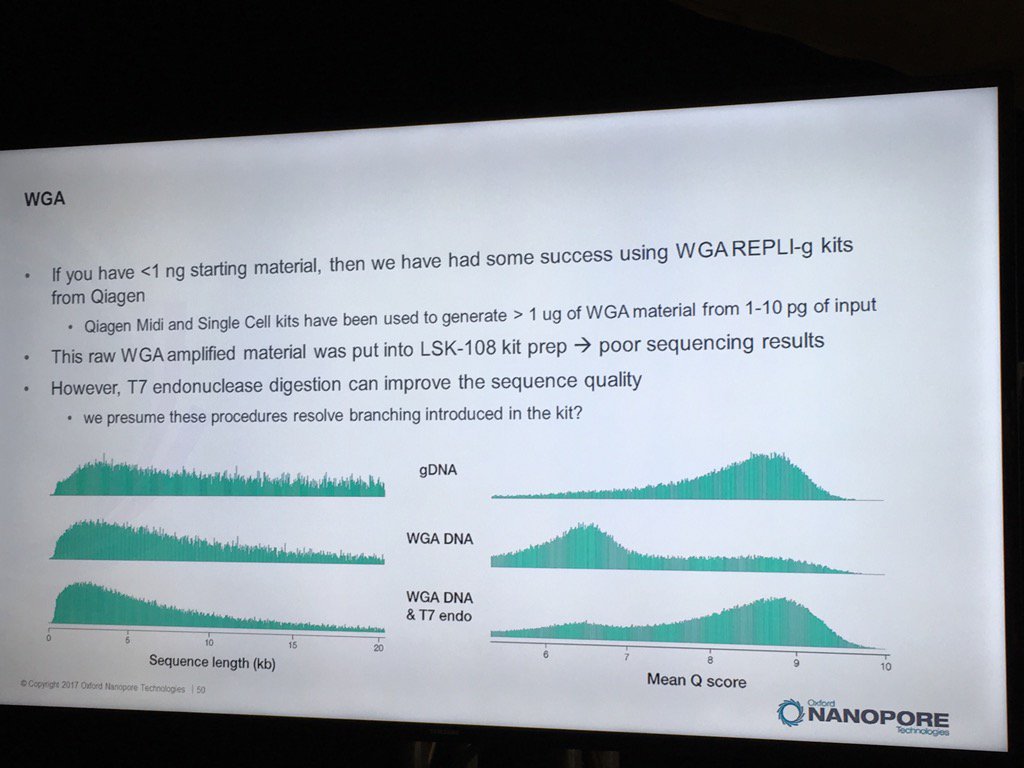
Oxford has had some good preliminary results using the REPLI-g WGA kit to generate runs from less than 1 nanogram of sample DNA. Higher yields and data quality are obtained by using T7 endonuclease to resolve the hyperbranched DNA structures. Hyperbranching is the result of the Multiple Displacement Amplification (MDA) reaction, in which a DNA strand may have multiple partially hybridized strands attached to it (each one having been partially displaced), which in turn can have partially displaced strands and so forth. The endonuclease treatment resolves these into a set of simple double-stranded fragments.
New MinKNOW Coming: A Few Glimpses
A new version of the MinKNOW software will be arriving soon, with a few features mentioned. This version is more attuned to controlling multiple flowcells on the GridION X5 and PromethION. Also, it will have more QC features. For example, the current "Platform QC" will be renamed "Check My Flowcell" and the output will be simplified to a "traffic light" scheme (upon which I triggered some Twitter discussion on the criticality of making such displays color-blind friendly, such as having both color and shape convey the status -- which the display does). Red is a bad flowcell; yellow are flowcells below the warranty specification but usable with that caveat and green are above spec flowcells.

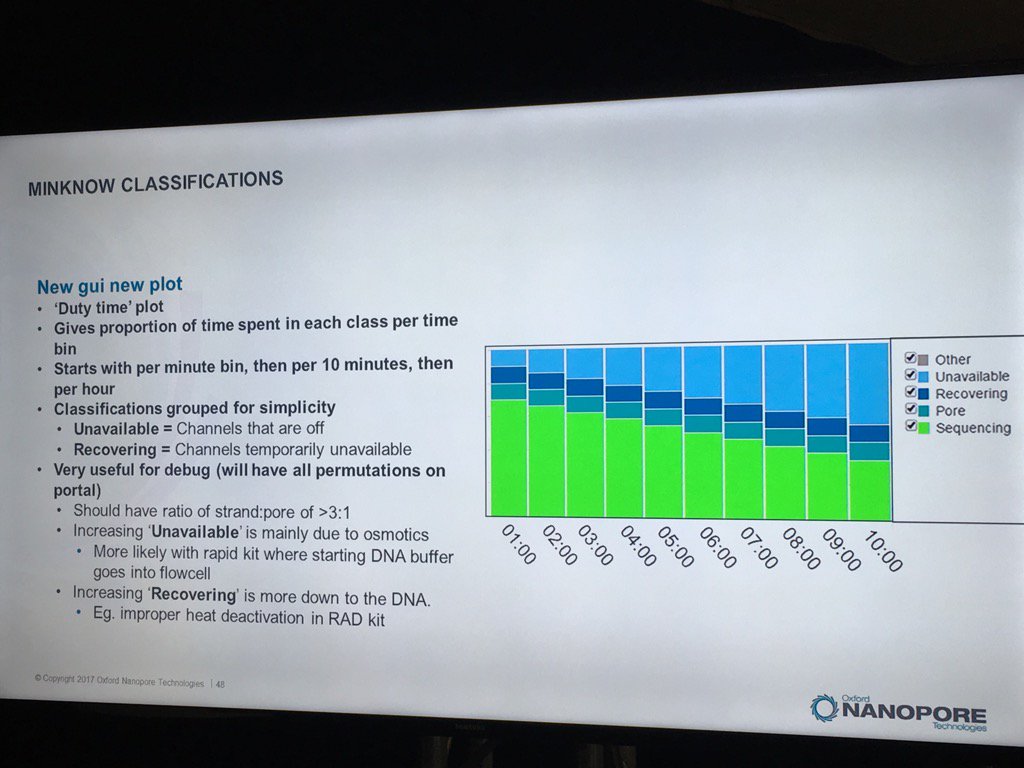
The new MinKNOW will have a display of the states of pores (compressed over the current displays) versus the time of the run. I don't see mux changes mapped on here, but this should be helpful in seeing the trajectory of a run
The existing bad categories were explained in further detail (you can also get this via a mouse-over in MinKNOW of the status display, a feature I suggested). Unavailable are pores that may be salvageable by the system reversing the voltage to try to clear a jam. Saturated are forever dead due to a popped membrane, multiple are useless due to having more than one pore and zero have no current flow. A spatial zone of pores in zero state can indicate a bubble on the sensor array. The "10 pA to inf" is a catch all and apparently can include pores that have just captured a strand.
The support for multiple flowcells will include the ability to group them and report aggregate statistics for groups.
Miten Jain on Long Reads
Miten Jain from UCSC gave a nice overview of the evolution of long reads on the platform. With his "Longboard" protocol, they routinely convert BAC preps into runs with read N50s of 130-150Kb, which means more than half the data is in full-length BACs that have been cut once by transposase. His recipe for this is
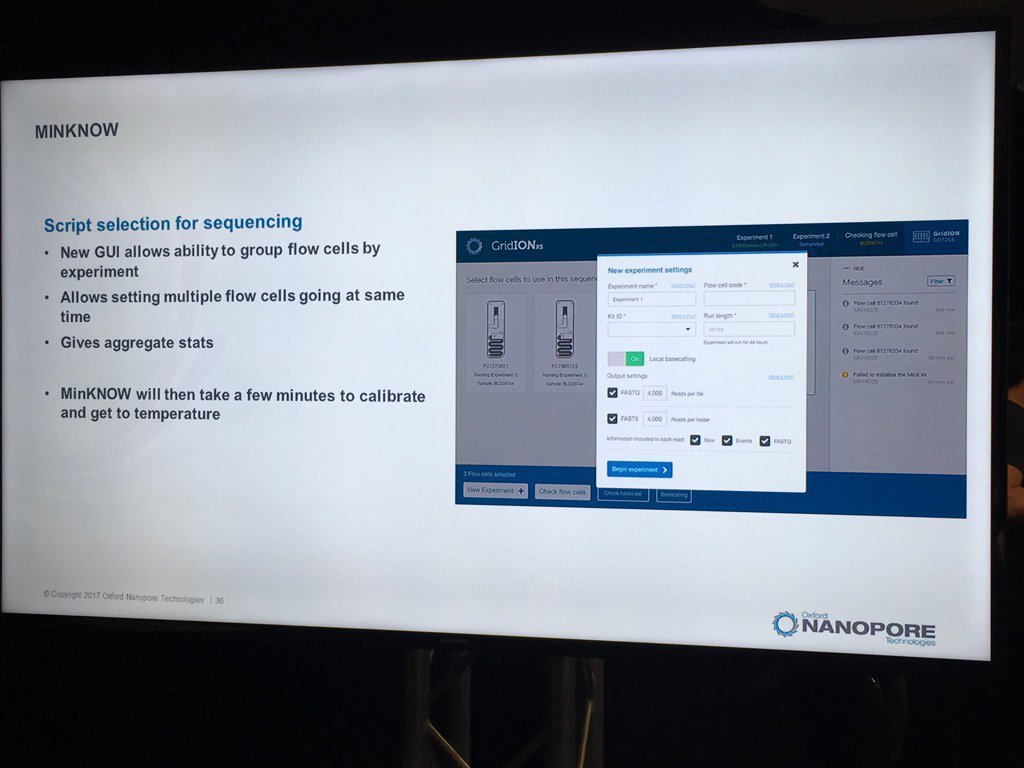
Miten warned against using SPRI beads with long DNA. In a ligation-based experiment he ran using the standard HindIII lambda digest spiked with an equimolar amount of full-length lambda, a sharp dropoff was seen in the detection of intact fragments by the sequencer, with no full-length reads recorded.
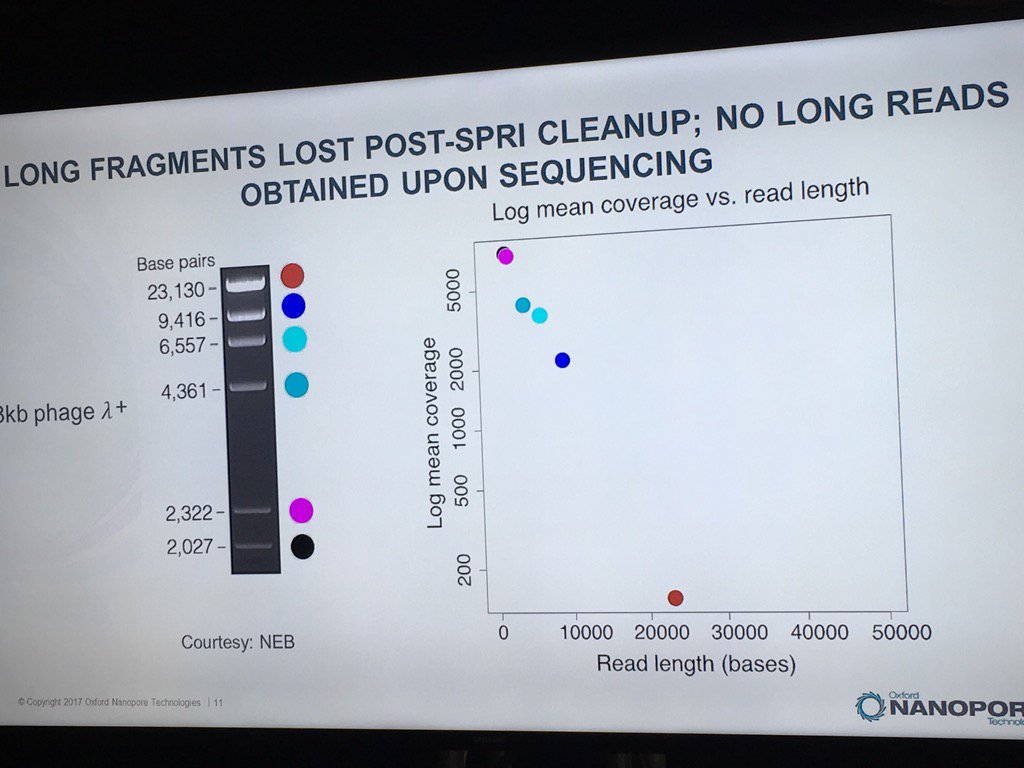
Miten started his talk by pointing out the disparity of scale: if the flowcell membrane is represented by a 1cm wide Sharpie pen, then a 180 kilobase fragment is like the London Shard tower, which conveniently looms across the river from the venue.
Miscellanea
Platform QC (the soon to be "Check My Flowcell") was explained a bit. There's really three stages to it: a 10s triangular wave to assess membrane integrity, a 30s of 180mV to test for single pores and then the sequencing of small, defined DNA fragments in the storage buffer to assess functional pores.
Hans Jansen gave a bit of a speculative talk (particularly for a workshop mostly full of novices) about future directions his group is taking their TULIP assembler and the challenges of assembling the tulip genome, as each chromosome is essentially one human genome in length. He did have a nice slice illustrating how N50 statistics or other gross assembly metrics can be less than fully helpful: despite the decent distribution of scaffold sizes below, the king cobra genome via short read technologies had terrible contiguity in the most interesting part, genes for venom components.
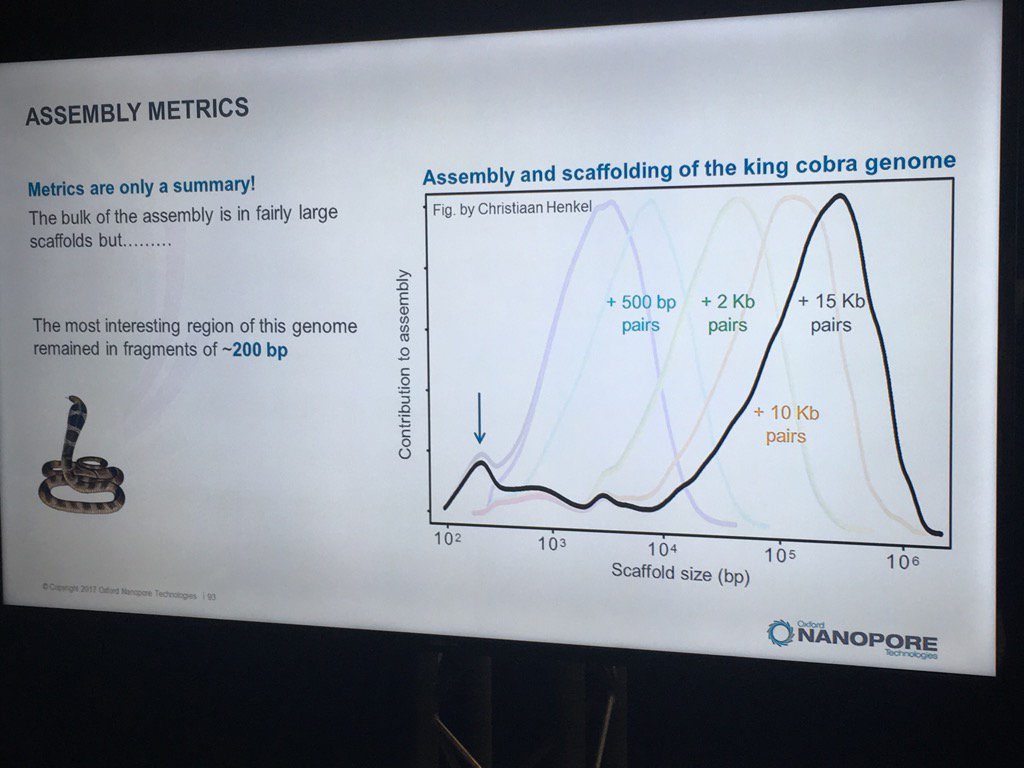
Jansen did finish with the thought that one day researchers will look back on the idea of genome assembly and perhaps laugh, as chromosomes will simply be read end-to-end. For some smaller eukaryotic genomes, that premise isn't far off given the leviathan reads recently seen -- and I expect to hear much more about during the main conference.
The main event starts in less than an hour!
4 comments:
I am currently taking an online genetics course and it has occurred to me that having a nanopore sequencer lab would be such a very very cool addition to the course.
Oxford has the Minions costed at $1000. The uni would have no problem with that.
Perhaps they would buy 20 or 30 and send them out to the students for a 2-3 week loaner. The capital price depreciation would work fine for them.
Only sticking point would be the flow cells. I don't think they could budget $500 per flow cell per student. Would Oxford have miniflows available? Perhaps more like a Gbp? A flow that could be priced more like $50 would be within the budget of our uni and probably a bunch of other educational institutions. Could always try the flows that did not pass QC. As long as the lab work with flow cells was not overly technical I am sure doing nanopore chemistry would be greatly enjoyed by my classmates.
Hey you have great writing skills, apart from keen observation.
All the best
Raja Mugasimangalam
I was dying to go to the LC17, and especially the workshop, but couldn't, so thank you so much for posting this!
I think they now do have 'mini flow cells' they call 'flongle'. $100 each.
Post a Comment