A year ago, Ion Torrent was the least expensive, rapid turnaround sequencing system around, and we ran a number of genomes on the PGM. However, while it was useful to get a first look at a genome, the notorious homopolymer problem,. coupled with the limited number of assemblers which do well with Ion data (let alone are tuned for it), always led to frameshift-rich assemblies. Ion also has failed to get much traction in the academic bioinformatics community, which means there is not a rich set of open source tools for improving and cleverly filtering reads. Once MiSeq became accessible through service providers, I threw my allegiance solidly behind it.
However, hope springs eternal and the Ion basecaller should be improving. Plus, I had seen some hints that Ion data could sometimes bridge low coverage areas of Illumina assemblies, though the effect was all-too-rare and rarely where I needed it most. Still, with the advent of 300 basepair reads on the Ion, perhaps it was worth retrying the platform.
One of the samples sent for Ion sequencing was a Streptomyces genome. Now, that's my bread-and-butter, as most natural product drugs have come from these bugs and their kin (starting with streptomycin; if it has "mycin" or "micin" in the name, it's almost certainly from one of these). Streptomyces have many interesting properties for the genome sequencer, starting with their high GC content.
Now, a word about high GC content. When I did my first Sanger sequencing experiments, way back in my Delaware days, I was trying to sequence Chlamydomonas. It was notorious for high G+C -- somewhere i the high 50s/low 60s. While that can cause trouble for Sanger, that's hardly high; my standards have tightened a bit. A recent comparison of benchtop sequencing platforms used Bordetella pertussis as their high GC sample, averaging a bit over 2/3 G+C. Local sequences can go much higher, which makes B.pertussis a good start. As you are about to see, it is not a high G+C organism. I'm not claiming Streptomyces take the crown, but I think the plots below will convince you that B.pertussis is a piker in this department.
Since a major goal was to try out longer reads, I immediately plotted the distribution of read lengths. Yes, this was in the Ion quality report, but with a severely compressed Y-axis. The result is below, which was immediately striking.
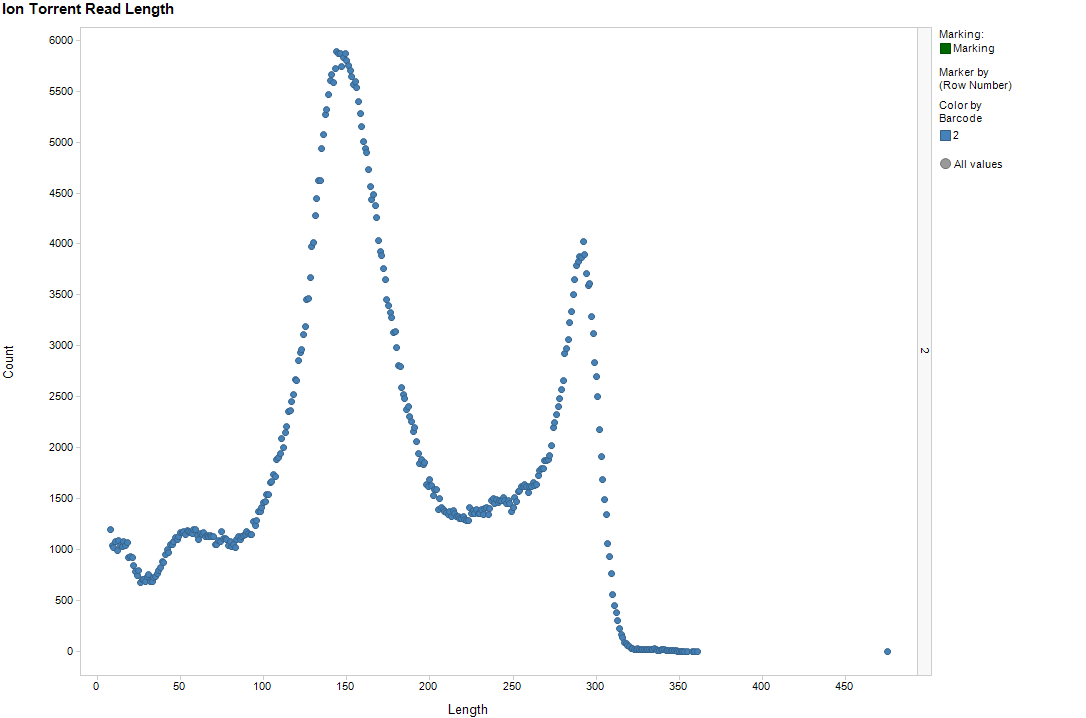
Okay, given the nature of the Ion chemistry, I wouldn't expect everything to line up in one position. A sloppy peak would not be surprising -- but such a strongly bimodal distribution?? That was a surprise.
Now, the same run actually included some other non-Streptomyces samples of normal GC content, and they showed only the long peak, not the shorter one. That was immediately a hint that GC content might be a variable worth looking at. So, I went next for that, trellising the data by GC content of the read.

A bunch of things to point out. As guessed, the long peak has a dependence on G+C, disappearing with the very long reads. There's that weird bit of looking like I'm plotting two data series; there's only one, so I'm guessing that I'm seeing some preference of the basecaller to go for certain lengths.
Finally, note where the bulk of the reads are, or more importantly how minuscule the fraction of reads are at 68 and below. Bordetella pertussis: PFFFFT! It's also worth noting the paucity of reads greater than about 77 G+C; this is quite possibly a dropout of the very high G+C reads. I haven't looked at windows this long, but in 50 basepair windows I've quit being shocked by regions of greater than 90% G+C. Like I said before, sequencing in these bugs is interesting.
4 comments:
Hey, whatever happened to Life's Grand Challenges or was that another
life joke....
This blog has become nothing more than a marketing subterfuge. Let's get back to the science and leave the advertisements to the rest of them.
Go back to your worm hole. Keith gives what I'd say is one of the more unbiased views of what is happening in the NGS space. He is beyond reproach...he tells it like it is...good and bad.
Nice blog :)
Post a Comment