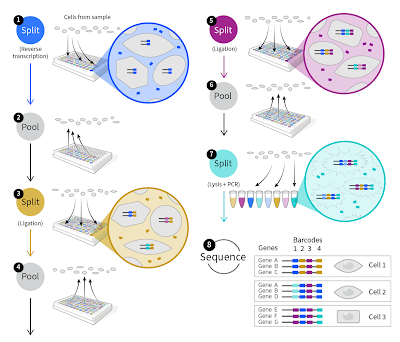
Just to review, the key concept (Split-Seq) in Parse' technology is that once molecules are linked to cell or nuclear membranes, these particles can be moved around. In the initial first strand cDNA synthesis the samples are tagged by well location and then be mixed. Subsequent labeling steps involve randomly splitting the pool, adding labels to each split of the pool, then re-pooling the mixtures. With sufficient splits, each cell's contents will accrete a set of barcodes specific to that cell.
Parse's "Evercode" kits come in three sizes: Evercode Whole Transcriptome Mini for trials and pilots and capable of tagging 10K cells total in up to 12 samples, Evercode Whole Transcriptome for up to 48 samples and 100K cells and Evercode Whole Transcriptome Mega targeting up to 96 samples and one million cells per experiment. Each kit has a 30 minute fixation step after which samples may be store indefinitely; this freezes the gene expression state. We discussed the advantages of this for long timecourses, situations such as getting clinical samples from surgeries or just waiting for a set of experiments to complete -- by quenching each sample and then running together one can minimize batch effects. Output of the process is a ready-to-load Illumina library. Each kit also comes with Linux software to process the data into standard formats which can feed into a variety of open source single cell expression analysis tools and a set of web-based reports running some basic clustering.
The system works well with isolated nuclei too. Nuclei offer a number of advantages, particularly in terms of working with solid tissue. Disaggregating cells often uses enzymes that must work at relatively warm temperatures -- and the cells may be shifting their expression in the meantime. Nuclei preparation procedures are typically run a cool temperatures, slowing changes in expression patterns. Of course nuclei will have incompletely processed messages, but apparently what is there is sufficiently representative for most analyses.
Not having analyzed any single cell experiments myself, I asked how much data does one need? It of course depends on the complexity of the transcriptome and how sensitive one wishes to be for low expression transcripts, but ranges of 5K reads per cell to 25K or 50K are common. Don't go too crazy: if you really wanted to go deep on 1M cells, that would be 50B reads or about five NovaSeq S4 flowcells (at 8-10B reads passing filter). Experiments that mongo might remain a glimmer in an Illumina salesperson's dreams for quite a while!
The kits use a combination of oligo dT and random hexamers to prime first strand cDNA synthesis. While the random hexamers increase the burden of ribosomal RNA sequenced (perhaps 10-15% of reads), Parse finds that it significantly increases the number of rare transcripts detected and evens out read coverage across each transcript, for an overall win
Overall, the workflow is about 2.5 days, which represents a significant streamlining from the original publication in 2018. It's also relatively automation friendly thought the founders assure me that even someone "straight out of undergrad" can master the pipetting steps (hmmm, I could once pipet competently...)
Parse plans to use the new funding to greatly expand on all fronts; if you like a supervolcano looming in your backyard they plan to do a lot of hiring. Some of this will be used to expand their commercial presence - currently they cover US, Canada and Europe themselves and have started partnering for distribution in other areas - but they also plan to push in a number of technical directions.
For example, they see great promise in multi-omic profiling; a wide variety of molecules can be attached via fixation to cell membranes. We didn't go into details, but it is easy to imagine oligo-linked antibodies being used for detection. Immunoprofiling and capture-based methods are also on their radar. They haven't run any long read experiments, but customers have been pairing the Parse technology with PacBio and Oxford Nanopore
While the current offering will focus on mammalian systems, there have been publications using the underlying Split-Seq technique for various microbes, including bacteria. Both for day job reasons and just general love of bacteria, that's very exciting to me -- there has been a dearth of single cell expression experiments on microorganisms but certainly there isn't a lack of interesting biological phenomena which could be studied with such experiments.

Parse founders Charlie and Alex
(all photos courtesy of Parse Bio)
No comments:
Post a Comment