Okay, some ground rules and such. I'm not a professor and I don't get paid to teach, nor do I have much experience with it. Doing some part-time teaching is up there with being a dog walker at a boutique hotel in my list of fantasy retirement jobs, but I'm not planning on retiring anytime in the forseeable future. I do recognize that teaching well requires real effort, and that good coursework tends to be a work in progress. Perhaps what I outline here is too ambitious, but I tend to think not -- but how would I know since I don't teach?
Related to that subject is a key ground rule: I do not believe in spoon-feeding! Among the core capabilities one needs in the field are:
- Use Google to find answers to basic questions
- Use Google to find code repositories on GitHub and others
- Use PubMed, BioRxiv and Google to find literature related to a topic
- Reading README files
- Having the patience to figure out if a program will kick out a summary of commands if run with no arguments, or with -h, -help or --help as arguments
And so on. If you run into a snag, first try one of these. Then try the rest of them. Then maybe ask for help.
Also, I'm spoiled on compute power. I think everything I propose here can be done on a typical laptop or the free tier of Amazon, but you might need to be a bit patient. I haven't run through any of this; the concept is sound, but execution is entirely left as an exercise for the student. The beauty of computational biology is that so much can be done on consumer hardware, Amazon free tier or public servers -- but perhaps this idea is beyond those limits.
It would be fun to actually do this with a group of motivated students, as I could imagine breaking the problem into sub-problems, with different students/teams tackling different partitions. Or having different students try different approaches to solve the same high-level problem.
Does Transcription-Translation Coupling in Bacteria Leave a Signature in Sequences?
So what is the result that got me going on this? A brief paper in Science provided a structural biological solution to a long-standing question: just how tightly tied together are transcription and translation in bacteria? Eukaryotes separate these two processes: transcription in the nucleus, translation in the cytoplasm. But in bacteria, it is well-known that ribosomes don't wait for RNA polymerase to finish before they start translating. This is the basis for some interesting regulatory schemes such as attenuation, in which whether translation pauses or not determines where RNA polymerase terminates transcription. The new paper uses cryo-electron microscopy to show an intimate interaction between RNA polymerase and the ribosome. Now cryo-EM is often not a hyper-detailed look; a former colleague who had done some warned that you can be trying to fit known crystal structures somewhat ambiguously into blobs of electron density, but the derived model shows how the two fit together.
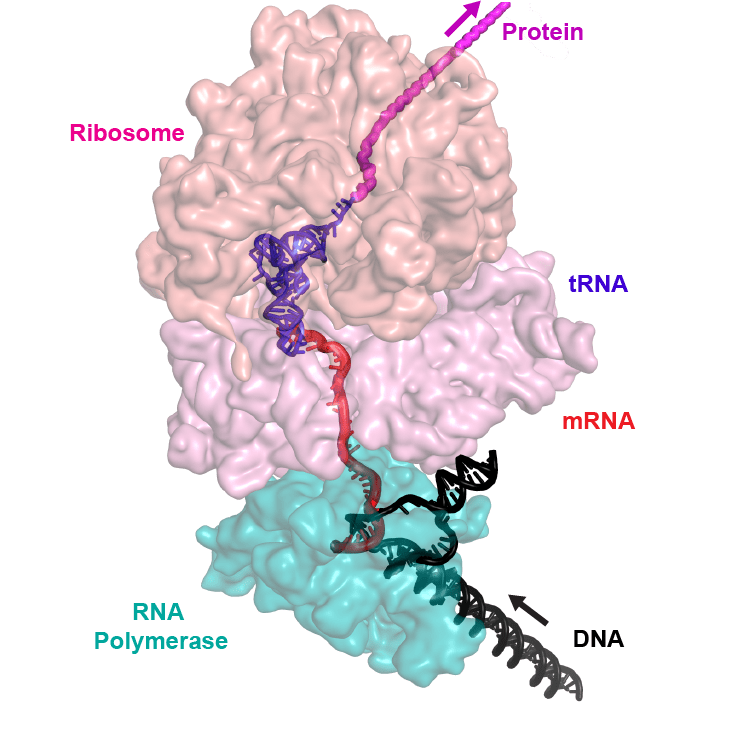 (image from University of Wisconsin publicity article, where the work was performed)
(image from University of Wisconsin publicity article, where the work was performed)
So here's the question? Given that ribosomes and RNA polymerase are closely docking, can we find evidence for that in sequences of ribosomal proteins and RNA polymerase subunits? Do that that, we'll need to dive into the expanding world of sequence co-evolution analysis.
Detecting Co-Evolution, A High-Level Overview
Suppose two proteins have been interacting with each other over very long evolutionary times. It might stand to reason that should an amino acid at the interface mutate in one, then there might be selective advantage to a mutation in the other to compensate. Or certain mutations would simply not be tolerated as they would reduce fitness; if the interaction relies on a salt bridge then one partner might always need to maintain a positively charged side chain and the other a negatively charged side chain.
Co-evolution analysis looks for these patterns in deep protein multiple alignments. Really, really deep protein multiple alignments. Perhaps 10K, 20K or deeper multiple alignments. After all, there are 400 possible amino acid pairs, so we'd like the alignments to be deep enough that we will sample a reasonable number of allowed pairings to estimate their compatibility. Plus we're going to be comparing every possible pairing of positions, which means we have a very large N. It would be good to have very deep alignments to identify highly coupled positions, given that there will be a huge background of uncoupled and weakly coupled positions.
There is a quirk to this analysis that is worth emphasizing in case you've worked with multiple alignments before. It is easy to get excited in a multiple alignment over a position which is perfectly conserved, because that tells you that a specific amino acid is somehow crucially required for function. Conversely, one tends to ignore positions that show high variability; perhaps these are in loops where the sequence doesn't matter.
But with co-evolution analysis, what we are roughly looking for is mutual information. In other words, given amino acid AAi at position i in the alignment, do I have any ability to predict amino acid AAj at position j. If I do, then those positions are coupled. But if a position is perfectly conserved, I can't possibly make a prediction of another site based on that site: the first one is always the same! Conversely, two sites could be each apparently unconserved but have a strong coupling between them.
There is clearly another issue: transitivity. If amino acid position B is located in space directly between amino acid positions A and C it may well be coupled with both A and C, but it also might appear that A is coupled with C. Presumably the A-B and B-C relationships will be numerically stronger than A-C, but if we don't already know the spatial arrangement we might not realize that the A-C relationship is due to transitivity. Software in this space may try to detect and filter out such transitive relationships, but in any case one should keep that possibility in mind when interpreting findings.
There is clearly another issue: transitivity. If amino acid position B is located in space directly between amino acid positions A and C it may well be coupled with both A and C, but it also might appear that A is coupled with C. Presumably the A-B and B-C relationships will be numerically stronger than A-C, but if we don't already know the spatial arrangement we might not realize that the A-C relationship is due to transitivity. Software in this space may try to detect and filter out such transitive relationships, but in any case one should keep that possibility in mind when interpreting findings.
Since we are dealing with biological sequences, and not those spit out by a Hidden Markov Model or similar algorithmic generator, there is an additional complication: phylogeny. The ability to predict AAj given AAi can reflect a physical interaction between those positions, or it could reflect common history. Some approaches in this space see phylogeny as a bug, and try to wash it out mathematically, whereas others see it as a feature to enhance sensitivity. Know which one you are using!
This sort of co-evolution analysis has been run on a grand scale in a recent paper from David Baker's group, in which they predicted three dimensional structures for hundreds of proteins of unknown function, using metagenomic databases as a rich source of additional alignments. And I do mean predicted structures: the coupling values derived from the multiple alignments can be treated similarly to distance constraints from NMR data and used to generate actual structures.
The Workflow
Here are the key steps I envision. Now each of these steps can be simple or complicated, and there are a number of interesting side-excursions. They also touch on many highly relevant issues in bioinformatics, such as workflow management, format conversion, tracking multiple discontinuous coordinate systems, and visualization.
Generate multiple alignments for ribosomal proteins and RNA polymerase subunits
The first step is to create the required multiple alignments. A potential catch here is that many ribosomal proteins evolve remarkably quickly, so a trivial BLASTP search might not find everything. Then again, if our goal is to generate really deep alignments, then making sure BLAST returns all the the hits can be tricky. PSI-BLAST, which iterates is one solution. Using HMMER to search Hidden Markov Models (HMMs) is another. There is also the question whether to follow the path blazed by Baker's group and go into metagenomic resources. And you'll need a multiple alignment program which can handle really deep alignments.
But after you've collected huge numbers of proteins, there is potentially a problem on the other side. Some taxa have been sequenced to extreme depth, and if your 20,000 deep alignment has 5,000 E.coli sequences in it that's going to skew the results. So some sort of protein clustering followed by dataset reduction will be in order. There are many ways to do this: start with pre-clustered databases, collapse identical sequences, full-blown clustering.
Generate Coupling Scores
There are a number of packages out there to generate co-evolution scores. Read the directions carefully! There is a snag which may well show up here, and you need to be prepared. Some of these packages have a hard-wired limit on the width of multiple alignment they can be fed. If you want to look at two really large proteins, you might blow out this limit. A strategy I have used is to prune out columns that are gapped in most of the sequences. Since these programs are just comparing columns, that doesn't affect the results. However, it does mean that you need to track these omissions so that you can map back onto known structures.
Also, as you may have picked up, these programs look at a single multiple alignment. If you want to look for coupling between positions in two different proteins, then you need to create a multiple alignment that concatenates the two proteins, carefully maintaining each row as one biological species. In other words, row 1 has E.coli ribosomal protein S7 and E.coli RNA polymerase subunit beta, row 2 has B.subtilis ribosomal protein S7 and B.subtilis RNA polymerase subunit beta, etc. So in all your multiple alignments you need to keep this pairing information handy. Perhaps a good time to write a simple SQL database using SQLite?
Visualize on Crystal Structures
Once you generate all those coupling scores, you'll want to filter them to find the strongest interactions. Perhaps compare the positions found ribosomal proteins with those already reported for ribosomal proteins, to check if your cutoffs are appropriate. Once you have some interesting results, there needs to be a way to visualize them.
The best way, since we're trying to look if 3-dimensional interactions have left a signal in the sequence, is to show where the interacting residues are on the crystal structures of the component proteins. PyMol is a great tool for that, with its own scripting language which allows complete control of rendering. So you could color the regions by strength of coupling, show the coupled positions as space-filling models while the rest of the protein is a ribbon diagram or what have you. But this is why I warned above about keeping track of coordinates: your gappy, perhaps pruned multiple alignment needs to be mapped onto the protein structure you are using for visualization.
Visualize the Coupled Positions
This is perhaps the least well-worn part of this proposal, but an interesting follow-on. If you find coupled positions, perhaps lots of them, what can you say about them from a sequence perspective? Imagine collapsing the alignment columns for any pair i,j into a 40 element vector, where the first 20 positions have the frequencies of amino acids at position i and the second 20 the frequencies at position j. Now feed the matrix of such vectors into a two-way hierarchical clustering program. On the one dimension, you'll find the positions clustered by similarity in their amino acid patterns. Are there recognizable patterns to any of these such as probable salt bridges (opposite charges between the i and j amino acids)?
Of course, you could take the actual coupling matrix and cluster it as well -- 400-long vectors capturing the propensity of observed frequency for each possible AAi,AAj. That will be a much sparser matrix and perhaps the result is uninteresting -- but how will you know unless you try it?
Does That Look So Hard?
So there you have it: a complete, biologically relevant analysis in four (not so) easy steps. And if that isn't enough, well one could build trees from all those alignments. Or one can see if patterns discovered in the entire dataset seem to hold true for subsets. For example, does it look like chloroplast and mitochondrial transcription-translation systems retain the tight coupling of their bacterial forebears? The alignments of these subsets are probably too shallow to do the analysis on their own, but given the patterns found in a large bacterial dataset it should be possible to score these using some sort of function derived from the large dataset.
If you want to start with something simpler, just look for coupling within some of these protein families. That's a good warm-up to the whole beast but eliminates the need to carefully glue together multiple alignments.
If you want to start with something simpler, just look for coupling within some of these protein families. That's a good warm-up to the whole beast but eliminates the need to carefully glue together multiple alignments.
If this isn't to your tastes, there's plenty of other ideas I have for student projects. For example, there are all sorts of analyses that could be run on public human genome sequences, looking carefully (for example) at specific types of errors. Doing some simple text-mining on PubMed.
No comments:
Post a Comment